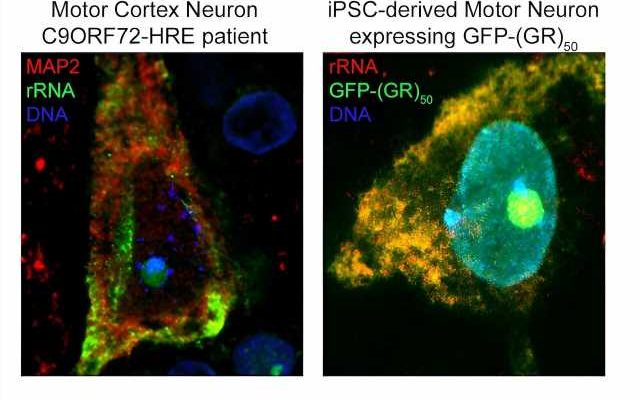
50 lentivirus, and stained for rRNA, showing the total overlap of poly-GR and rRNA in human motor neurons. Credit: Juan Ortega, Ph.D.")
A pair of studies from the laboratory of Evangelos Kiskinis, Ph.D., associate professor in the Ken and Ruth Davee Department of Neurology’s Division of Neuromuscular Disease and of Neuroscience, have uncovered novel cellular mechanisms that are involved in two types of genetic amyotrophic lateral sclerosis, or ALS.
The findings, published in Science Advances and Cell Reports, improve the understanding of ALS, a progressive neurodegenerative disease that attacks motor neurons in the brain and spinal cord, and provides support for the future development of targeted therapies.
‘Baiting’ genetic mutations
An estimated 32,000 individuals are currently living with ALS in the U.S., according to the Les Turner ALS Foundation. There are two types of ALS: sporadic (non-genetic), which makes up more than 90% of all ALS cases, and familial (genetic).
In their paper published in Science Advances, the Kiskinis team studied a type of familial ALS caused by a repeat genetic sequence of the C9ORF72 gene, which is the largest genetic cause of ALS.
According to Kiskinis, healthy individuals may have 20 repeats of C9ORF72 in their genome, while patients with ALS can have thousands. These repeats are transcribed and translated through non-canonical pathways which produce irregular RNAs and dipeptides (molecules with two amino acids bonded by a peptide) in neurons.
Kiskinis’ team hypothesized that this cascading effect is what leads to gain-of-function toxicity in motor neurons in this type of ALS. This prompted them to investigate the mechanisms that make these repeated dipeptides (R-DPRs) increasingly toxic.
Using computational and experimental techniques, the investigators found that R-DPRs have a strong binding affinity for RNA molecules. Then, they used a technique called crosslinking immunoprecipitation, or CLIP-seq, to isolate specific RNA fragments and found that R-DPRs bind exclusively to ribosomal RNAs. Specifically, the poly-GR dipeptide binds to ribosomal RNA, which impairs ribosomal homeostasis, a process essential for cell differentiation and the overall makeup of a cell.
Using these findings, the investigators then designed an RNA “bait,” which tricks poly-GR into binding with something that appears to be ribosomal RNA —or in this case, the bait. In both in vivo models and in iPSC neurons derived from patients with C9ORF72 mutations, the bait molecule inhibited toxicity.
“We showed using multiple different approaches that the molecule we designed—the ‘bait’ —binds very highly and specifically to this poly-GR protein, and in binding to it prevents it from binding to ribosomal RNA and prevents it from going into the nucleus, which is where it becomes most toxic,” Kiskinis said.
“It serves as a proof of principle to the idea that an RNA molecule that has these ‘bait’ capacities can be therapeutic and it also strengthens the hypothesis that a lot of the toxicity from the C9ORF72 mutation is associated with impairing ribosomal biology.”
The investigators are now optimizing the chemistry of the bait molecule and aim to test it in other models of ALS, according to Kiskinis.
“We’re excited by the fact that iPSC-derived neurons from patients that have these mutations survive a lot longer when we give them this bait molecule, which means that it’s doing something good. We just need to figure out if it could be a viable and transformative therapeutic,” Kiskinis said.
Linking causal genes
There are 30 known genes associated with familial forms of ALS, but whether each genetic driver contributes to just one general type of ALS or multiple types has remained a looming question in the field, according to Kiskinis.
To address this question, according to their study published in Cell Reports, Kiskinis’ team used patient-derived induced-pluripotent-stem-cell (iPSC) spinal motor neurons to develop a model of the second most common type of familial ALS, which is caused by mutations in the SOD1 gene and is apparent in two percent of all ALS cases.
“We know from work that’s been done in the field over the last 20 years, that this mutation causes toxicity through gain-of-function effects. We’ve known that mutations lead to protein misfolding, and the misfolding starts a cascade that leads to dysfunction. But how that happens we don’t quite understand,” Kiskinis said.
The investigators used temporal proteomics to compare the proteomes of control iPSC neurons and iPSC neurons with a SOD1 mutation and compared the rate of protein degradation (the recycling and replacing of old proteins) between both sets of neurons.
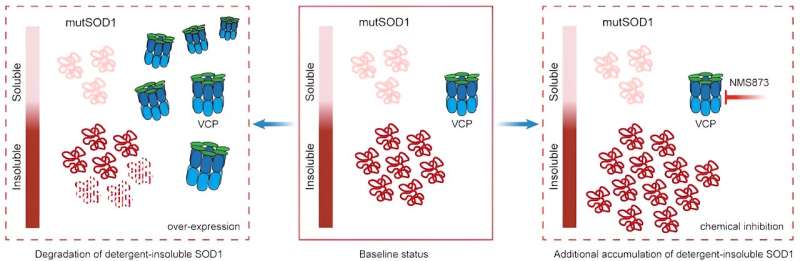
In the mutant-SOD1 iPSC neurons, they found that another ALS causal protein—valosin-containing protein (VCP)—degraded slower than in the isogenic control iPSC neurons. This slow protein degradation ultimately caused VCP to interact less with certain proteins and more with other proteins.
“This is quite exciting because mutations in VCP that can cause ALS are thought to act in a similar way,” Kiskinis said.
The investigators then aimed to determine whether this altered VCP function played a role in SDO1-mediated toxicity. In both mutant-SOD1 iPSC-patient neurons and C. elegans (a type of roundworm) models of mutant SOD1, the investigators found that when VCP was overexpressed, toxicity decreased and when VCP function was inhibited, toxicity increased.
“This paper, and work we’ve done in our lab over the last few years, are demonstrating that while the genetic causes might be different, there’s always some sort of level of mechanistic overlap when it comes to the causes of dysfunction in these cells,” Kiskinis said.
This is important, Kiskinis added, because the findings can inform the design of future clinical trials and improve the application of targeted therapeutics. Going forward, Kiskinis said his team wants to determine if VCP is an effective therapeutic target for patients with a SOD1 mutation, as well as for other types of ALS.
More information:
Juan A. Ortega et al, CLIP-Seq analysis enables the design of protective ribosomal RNA bait oligonucleotides against C9ORF72 ALS/FTD poly-GR pathophysiology, Science Advances (2023). DOI: 10.1126/sciadv.adf7997
Konstantinos Tsioras et al, Analysis of proteome-wide degradation dynamics in ALS SOD1 iPSC-derived patient neurons reveals disrupted VCP homeostasis, Cell Reports (2023). DOI: 10.1016/j.celrep.2023.113160
Journal information:
Science Advances
,
Cell Reports
Source: Read Full Article