
 insights from industryLaura NeaVP of Quality and Regulatory ManagementTecan
insights from industryLaura NeaVP of Quality and Regulatory ManagementTecanIn this interview, News-Medical talks to Laura Nea, Vice President of Quality and Regulatory Management at Tecan, about the upcoming changes to the regulations surrounding in-vitro diagnostics and how they might affect the life sciences landscape.
Video Credit: Tecan
Could you please give our readers a brief overview of your role and your background?
I am the Vice President of Quality and Regulatory Management at Tecan. I have worked at Tecan for over 10 years, and have over 30 years of experience in the IVD industry.
What is IVDR, and what is the timeline for its introduction?
The In Vitro Diagnostics Regulation (IVDR) is the European Union's complete regulatory overhaul of pre- and post-market requirements for IVD devices.
It expands the regulatory landscape to include the entire supply chain, not only in terms of manufacturers with responsibilities for design, development and commercialization but also in terms of agreements between manufacturers and key economic operators, such as importers and distributors.
IVDR also introduces specific requirements, and limitations on hospitals and labs developing their own diagnostic tests.
IVDR came into force on May 26th 2017, with a five-year transition period that ends on May 26th 2022. After that date, no new certificates under the previous IVD Directive (IVDD) will be valid.
There is a grace period that ends May 27th 2025, after which it will no longer be permitted to place an IVD product on the European market unless it is IVDR certified.
Image credit: Angellodeco/Shutterstock.com
What benefits does the IVDR bring to the market?
The benefits include improved quality and safety for all IVD devices, and an increase in post-market vigilance to ensure that devices remain compliant throughout their entire lifecycle, from cradle to grave.
IVDR also increases transparency throughout the entire supply chain, and outlines clear responsibilities between the economic operators, including manufacturers, distributors, importers and authorized representatives.
IVDR contains very specific requirements relating to both analytical and clinical performance, and every manufacturer must demonstrate that its devices meet these requirements.
There is also a set of general safety performance requirements (GSPRs) that set out very detailed requirements for device safety and quality.
What are the major changes in the new IVD regulations compared to the preexisting (and now being phased out) regulations?
The first key change is in device classification. There is a huge shift in how devices are classified between the previous directive and the new regulations.
Under the previous regulations, about 80 % of IVD devices were allowed to be self-certified by a manufacturer. Technical documentation was placed on the market under only the oversight of the manufacturer, and a notified body was not required to review that technical documentation in advance of the product going to market.
Under the new classification scheme in IVDR, only around 20 % of class-A products can self-certify, and about 80 % of devices will now need to be reviewed by a notified body. That notified body will not only review the technical documentation in advance of the product going to market, but it will also maintain oversight throughout the lifecycle of the product.
The definition of an IVD product has also changed to include more types of devices. The original directive was issued in 2001, and did not include some of the newer devices – such as genomic devices and personalized medicine – that are more prevalent today.
The technical documentation required to support the approval is also very detailed and prescriptive, requiring multiple plans and reports to support the scientific validity of the analyte, the analytical performance parameters and the clinical performance.
Currently, manufacturers can use legacy data – original data used when they brought the product to market under IVDD – but this may no longer be sufficient to meet the new requirements under IVDR. Additional studies may be required.
There is also comprehensive post-market surveillance in place for all IVD devices, including dedicated plans that require a manufacturer to update the technical documentation, ensuring that there is a continuous evaluation of new hazards and risks.
As technology evolves and global circumstances shift, there may be a need to update risk profiles, for example, to account for changes in antibiotic resistance, new interfering substances or variations in stability profiles. These changes often require active product performance follow-up studies.
The roles of economic operators and the supply chain are also clearly defined in the new regulations, requiring contracts and agreements between those partners to be updated.
Finally, commercial labs creating their own in-house tests are now defined as manufacturers, meaning they must also meet the requirements of IVDR. There is a health institution exemption involving slimmed-down requirements, but to be defined as a health institution, an organization must be serving EU patients. Industry and hospital labs are still waiting for guidance in this area.
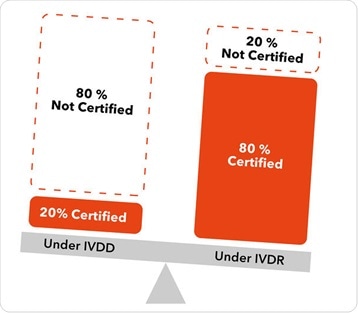
Image credit: Tecan
What are the implications of the new IVD regulations for manufacturers and other companies operating in the diagnostic space?
The IVD industry has begun to accept that there is increased responsibility for manufacturers to ensure all partners in the supply chain fulfill the new obligations. There is going to be increased oversight, and an uncertain future for companies that do not have the expertise or funding to comply with the new regulations.
IVDR has many more requirements and a greater level of oversight from the notified bodies, which will introduce additional costs for manufacturers to be able to market IVD products successfully under the new regulations.
Manufacturers will be forced to review and consolidate their IVD portfolios, potentially restricting the availability of certain assays or certain product types for a period of time.
Under IVDR, clinical labs will need to evaluate whether there is a commercial alternative for their in-house test. This means they will need to determine whether or not there is a CE-marked IVDR-certified device on the market that they could directly substitute for that particular in-house test.
There will also be implications for innovation and speed to market for novel diagnostic devices under the new regulations, but the scale of that impact still remains to be seen. Both notified bodies and the manufacturing industry are learning together how to correctly apply this new regulation.
Manufacturers will also need to assess the impact of the changes on current product lines that may be registered in other countries. For example, manufacturers may need to revise labeling to meet the new IVDR requirements, including any changes to intended purpose statements. These changes may require additional labeling or the resubmission of technical data to other countries where the product is registered, potentially duplicating that product with different labeling.
Finally, there will be a significant impact on the ability of notified bodies to absorb the additional workload and oversight requirements that will continue for the lifecycle of a product.
There are currently five notified bodies qualified to review IVD products under the new regulation. The situation can be thought of as a funnel, with many manufacturers with thousands of products passing through a tiny opening constricted by the limited number of notified bodies.
The tight timeline also factors into this bottleneck, as the majority of products will need to have moved through the IVDR process and be certified under that regulation by May 2022. The timeframe is short, the review times are long, and everyone is learning together. It is quite a challenge.
How will Tecan be tackling the IVDR regulation changes and what measures have already been carried out?
Tecan has embraced this challenge, and prepared for the new IVDR regulations from the outset. Our recommendation is to plan, engage and communicate.
The team at Tecan started planning for this transition upon publication, including quality system changes, reviews of supply chains for each facility designing and manufacturing IVD products, and undertaking a portfolio review and gap assessment of current technical files.
We engaged with our notified body early, including scheduling the required quality system audits, product classifications and groupings, and developing a submission roadmap.
It was also essential to communicate with management and internal stakeholders to ensure our understanding of the new requirements, roles and responsibilities, and manage expectations around costs, resources and timing. The increased requirements of a quality system should not be underestimated.
We were required to review and make hard decisions in our current portfolio of IVD products, deciding which ones to carry forward into the new regulation and constructing new technical files to meet the regulations. We developed a submission roadmap based on these new product classifications and groupings of our products.

Image credit: YanGe Tam/Shutterstock.com
Tecan was one of the first companies to gain IVDR certification, for our DHEA Saliva ELISA Diagnostic Assay Kit. Our experience was invaluable during this process, which highlighted the importance of the intended purpose statement and the impact of that statement on the classification of product and product groupings, and the clinical relevance associated with that product.
We have also had to update and expand the intended purpose statements for our products. For example, one of our IVD assay kits would have an intended purpose of ‘quantitative determination of vitamin D from human serum samples’. Under the new regulations, we were required to state how the kit is relevant or beneficial to a clinical condition.
In addition, requirements around our clinical performance data have been strengthened. They must now include at least one of three elements: prospective clinical studies, peer-reviewed literature or additional published experience gained by routine diagnostic testing, such as external performance and proficiency studies.
The landscape continues to change, and new guidance continues to be published. Each product submission will have its own challenges, so we conduct early reviews to look for gaps in technical documentation, comparing these to the new intended purpose statements and the requirements under the new regulations.
In terms of gaining the IVDR certification for the Saliva ELISA Diagnostic Kit, was this a hard process, or was it easier than Tecan was expecting?
There were elements that were difficult or unanticipated. We were able to learn from the interaction with the review process and the feedback from our notified body.
Some parts of the cycle – such as getting the quality system ready and upgraded to manage and pass the quality system audit – seemed fairly straightforward, because we already had robust quality control processes in place. Overall, it was very much a learning process.
What advice would you give to companies currently developing their strategy for the IVDR changes or companies that have yet to start even this preparation?
Preparation is key. Companies and labs need to engage with a notified body quickly, because time is running out and, if they have not started yet, there is going to be a delay. Notified bodies are already at capacity with current file reviews, and some have resource issues.
Companies need to take a critical look at their portfolios, and perform a gap assessment of any product that they want to transition to the new certification and market under the new regulations. This is essential to ensure that it truly meets all of the requirements of IVDR.
About Laura Nea
Laura Nea is Vice President QA/RA Management at Tecan. Laura and the multinational Global Regulatory team members partner with Tecan’s customers to ensure successful strategies for commercialization of products worldwide. Laura has over 30 years experience in the diagnostics and IVD market and has been on the Tecan team since 2010

![]()
About Tecan
Tecan is a leading global provider of automated laboratory instruments and solutions. Their systems and components help people working in clinical diagnostics, basic and translational research and drug discovery bring their science to life.
In particular, they develop, produce, market and support automated workflow solutions that empower laboratories to achieve more. Their Cavro branded instrument components are chosen by leading instrumentation suppliers across multiple disciplines.
They work side by side with a range of clients, including diagnostic laboratories, pharmaceutical and biotechnology companies and university research centers. Their expertise extends to developing and manufacturing OEM instruments and components, marketed by their partner companies. Whatever the project – large or small, simple or complex – helping their clients to achieve their goals comes first.
They hold a leading position in all the sectors they work in and have changed the way things are done in research and development labs around the world. In diagnostics, for instance, they have raised the bar when it comes to the reproducibility and throughput of testing.
In under four decades Tecan has grown from a Swiss family business to a brand that is well established on the global stage of life sciences. From pioneering days on a farm to the leading role our business assumes today – empowering research, diagnostics and many applied markets around the world.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices and treatments.